


 Новые взгляды на происхождение спинальной мышечной атрофии от Медицинского центра Ирвинга Колумбийского университета.
Новые взгляды на происхождение спинальной мышечной атрофии от Медицинского центра Ирвинга Колумбийского университета.
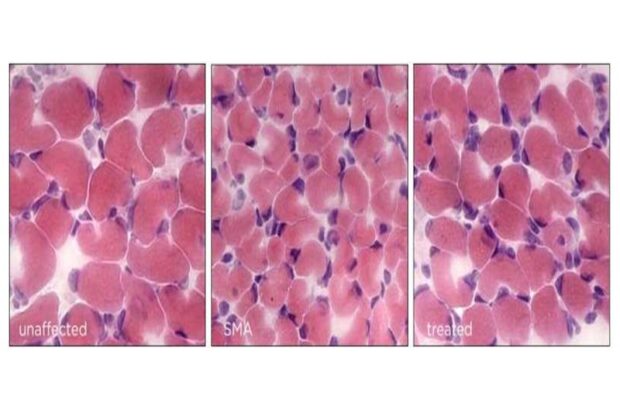
У мышей со СМА мышечные волокна восстанавливаются до своего нормального размера при изменении гена Hspa8. Предоставлено: Умрао Монани / Колледж врачей и хирургов Колумбийского университета Вагелос.
Исследователи из Колумбийского университета обнаружили, как генетический дефект приводит к спинальной мышечной атрофии (СМА). Эта важная информация о болезни, которую неврологи искали на протяжении десятилетий.
Открытие предлагает новый способ лечения СМА — разрушительного детского заболевания двигательных нейронов, которым страдает 1 из 6000 детей. В наиболее тяжелых случаях и при отсутствии лечения дети, рожденные со СМА, умирают в течение первых двух лет жизни.
Исследование опубликовано в журнале Neuron.
Исследователи также использовали свое открытие для разработки экспериментальной терапии, которая улучшила выживаемость мышей с тяжелой формой СМА в 30 раз, что является одним из самых высоких показателей, наблюдаемых при любом лечении на мышиных моделях СМА.
Почему открытие имеет значение
Почти все случаи СМА вызваны мутацией в одном гене — SMN1 (выживающий двигательный нейрон 1), который снижает количество белка SMN внутри двигательных нейронов . Дефицит белка SMN наносит вред нейронам, и в конечном итоге нейроны больше не могут контролировать мышцы тела.
СМА неизлечима, но обычно лечится с помощью терапии, которая увеличивает выработку SMN, включая генную терапию, которая вставляет новый ген SMN в двигательные нейроны.
«Для многих пациентов терапия довольно эффективна, если ее применять на ранних стадиях заболевания», — говорит Умрао Монани, доктор философии, исследователь СМА в Колледже врачей и хирургов Колумбийского университета Вагелос, который возглавлял исследовательскую группу.
«Но лечение работает не для всех, оно может иметь серьезные побочные эффекты, и, поскольку этим методам лечения всего несколько лет, мы не знаем, как долго они продлятся. Очевидно, что необходимы новые подходы».
Что обнаружило исследование
В поисках другого вида лечения команда Монани изучила мышей со СМА и попыталась выяснить, как дефицит SMN повреждает нейроны — то, что десятилетиями оставалось скрытым от исследователей, — чтобы найти способ предотвратить вред.
Исследователи обнаружили, что дефицит SMN обычно наносит вред нейронам, нарушая работу другого белка, Hspa8, который помогает установить важную связь между двигательными нейронами и мышечными клетками.
Когда Hspa8 не работает, коммуникационные каналы никогда не строятся, и сообщения не могут быть отправлены от нейрона к мышце. Мышцы не могут сокращаться, не получая этих сообщений, и в конечном итоге они истощаются.
Но команда заметила, что некоторые мыши со СМА были сильнее и менее поражены болезнью. Исследователи узнали, что у этих мышей был специфический вариант гена Hspa8, который не был нарушен дефицитом SMN.
Потенциальный новый путь лечения СМА?
Лечение, которое имитирует защитный эффект варианта Hspa8, может оказать сильное воздействие на людей, говорит Монани, если на это указывают эксперименты исследователей на мышах.
Используя подход, который преобразовал Hspa8 в его вариантную форму, исследователи обнаружили значительное восстановление нервно-мышечной функции и выживаемость у мышей с тяжелой СМА. Пролеченные мыши прожили около 300 дней по сравнению с 10 днями у нелеченых мышей.
«Мы никогда раньше не видели такого значительного увеличения выживаемости у этих мышей, даже при лечении нусинерсеном, текущим методом лечения СМА», — говорит Монани.
Необходимы дальнейшие исследования, чтобы определить, как лучше всего использовать полученные данные в новом лечении СМА. «Самый простой способ — инкапсулировать вариант HSPA8 в вирус и доставить генную терапию пациентам, как мы сделали с мышами», — говорит Монани. «Еще одна возможность — использовать вирусы или небольшие молекулы для превращения нормального Hspa8 в вариант».
В настоящее время исследователи работают над разработкой методов лечения, подходящих для тестирования на пациентах.
Дополнительная информация: Jeong-Ki Kim et al. Модификатор спинальной мышечной атрофии вовлекает белок SMN в сборку комплекса SNARE в нервно-мышечных синапсах, Neuron (2023). DOI: 10.1016/j.neuron.2023.02.004
Информация о журнале: Нейрон
Перевод https://nnd.name/
Оригинал: https://medicalxpress.com/news/2023-03-insights-spinal-muscular-atrophy.html



